CopyKAT Analysis
Preface
TIP
CopyKAT (Copynumber Karyotyping of Tumors) is an innovative single-cell CNV analysis tool, with its most prominent feature being the ability to automatically and unsupervised distinguish tumor cells (aneuploid) from normal cells (diploid) from single-cell transcriptomics data, and further resolve subclonal structures within tumors.
Unlike tools that require manual specification of normal reference cells, CopyKAT uses integrated Bayesian methods to automatically identify a group of cells with the most stable genome within the input cell population as internal references. This feature makes it particularly powerful in the following scenarios:
- Lack of clear normal controls: When there is no cell type in the sample that can be confidently identified as "normal".
- Exploring unknown tumor components: Automatically identifying potential malignant cell populations in complex tumor microenvironments.
- Simplifying analysis workflow: Enabling preliminary tumor/normal cell classification without pre-annotating cell types.
This document will detail CopyKAT's theoretical foundation, SeekSoul™ Online operation methods, result interpretation, and relevant application cases.
CopyKAT Theoretical Foundation
Core Principles
CopyKAT's core hypothesis is: In most tumor tissues, there are genomically stable diploid cells (such as immune cells, stromal cells) and genomically unstable aneuploid tumor cells. These two types of cells have systematic differences in their gene expression profiles at the whole-genome scale.
It identifies this difference through a multi-step algorithm:
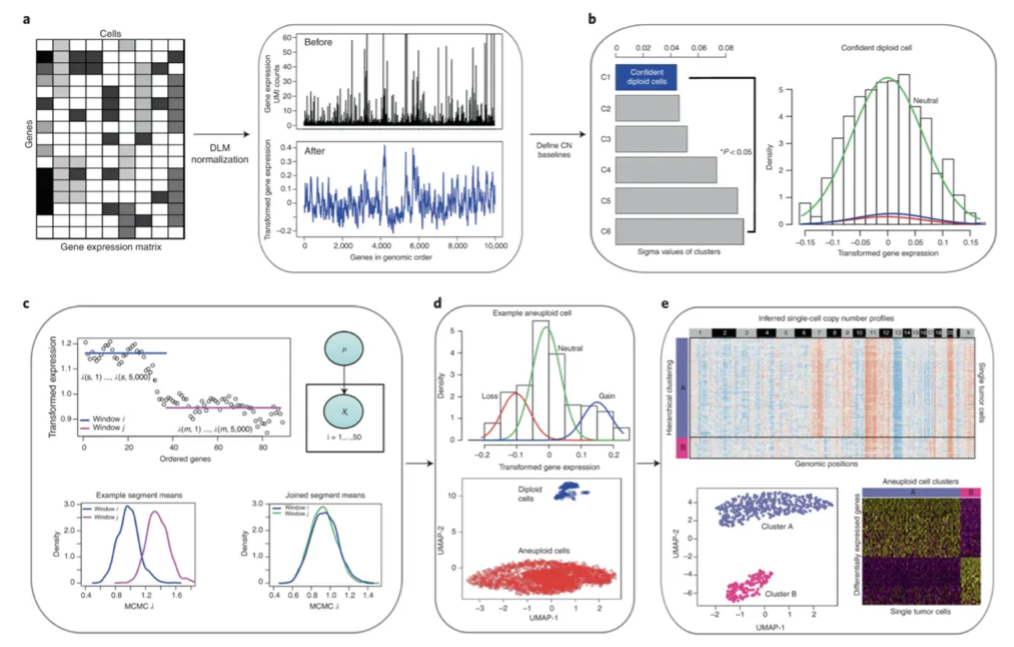
Analysis Workflow Overview
- Data Standardization: Normalize the input gene expression matrix to stabilize technical variance.
- Unsupervised Normal Cell Identification: Analyze the gene expression distribution of all cells using Gaussian Mixture Model to automatically identify a group of cells most likely representing diploid normal cells as internal references.
- Calculate Relative Expression: Using this group of internal reference cells as a baseline, calculate the relative gene expression level for each other cell.
- CNV Fragment Identification: Use Markov Chain Monte Carlo (MCMC) method to segment gene expression signals along chromosomes, identifying CNV breakpoints to define chromosomal segments with consistent copy number status.
- Cell Classification: Based on each cell's CNV profile, use Bayesian methods to calculate the posterior probability of belonging to "aneuploid" or "diploid", thereby classifying cells.
- Subclone Analysis: Within the tumor cells classified as "aneuploid", further divide them into tumor subclones with different CNV characteristics through hierarchical clustering of their CNV patterns.
SeekSoul™ Online Operation Guide
On SeekSoul™ Online, CopyKAT's analysis workflow is very straightforward.
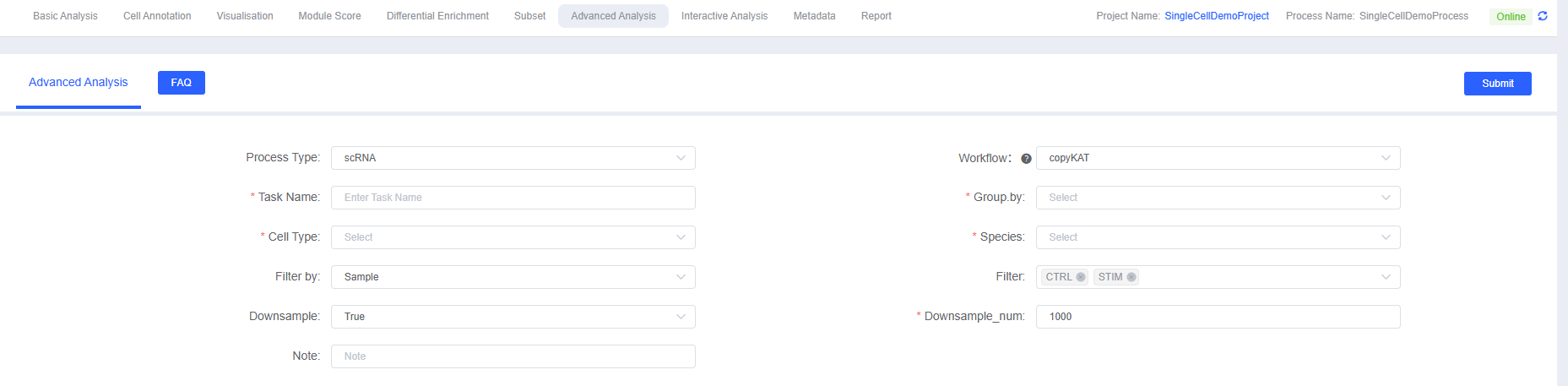
Parameter Details
| Interface Parameter | Description |
|---|---|
| Task Name | The name of this analysis task, must start with an English letter, can include English letters, numbers, underscores, and Chinese characters. |
| Group.by | Select the label corresponding to the cell type or cluster to be analyzed (such as CellAnnotation), used in conjunction with "Cell Type" below. |
| Cell Type | Multiple selection, select specific cell types or clusters to be analyzed (such as Epithelial cell, etc.). |
| Species | Select the species corresponding to the data, currently supporting human and mouse. |
| Filter by | Select labels used to filter samples or groups (such as Sample). |
| Filter | Multiple selection, select samples or groups to retain in the filter factor (such as Sample_A). |
| Downsample | Whether to downsample cells (extract part of the cells) for analysis. |
| Downsample_num | If downsampling is enabled, set the number of cells to be extracted here. |
| Note | Custom remark information. |
TIP
CopyKAT is very suitable for preliminary malignant/non-malignant screening of the entire cell population. You can select all cell types as input and let the algorithm automatically complete the classification.
Operation Workflow
- Enter Analysis Module: Navigate to the "Advanced Analysis" module on SeekSoul™ Online and select "copyKAT".
- Create New Task: Name your analysis task and select the sample or project to be analyzed.
- Configure Parameters: According to the above guidelines, select the cell type, grouping information, etc. to be analyzed.
- Submit Task: After confirming the parameters are correct, click the "Submit" button and wait for the analysis to complete.
- Download and View: After the analysis is complete, download and view the generated analysis report and result files in the task list.
Result Interpretation
CopyKAT's report intuitively displays the results of cell classification and CNV.
Cell Type Inference Heatmap
This is CopyKAT's core result graph, showing the CNV profile of all cells and the prediction of their "tumor/normal" identity.
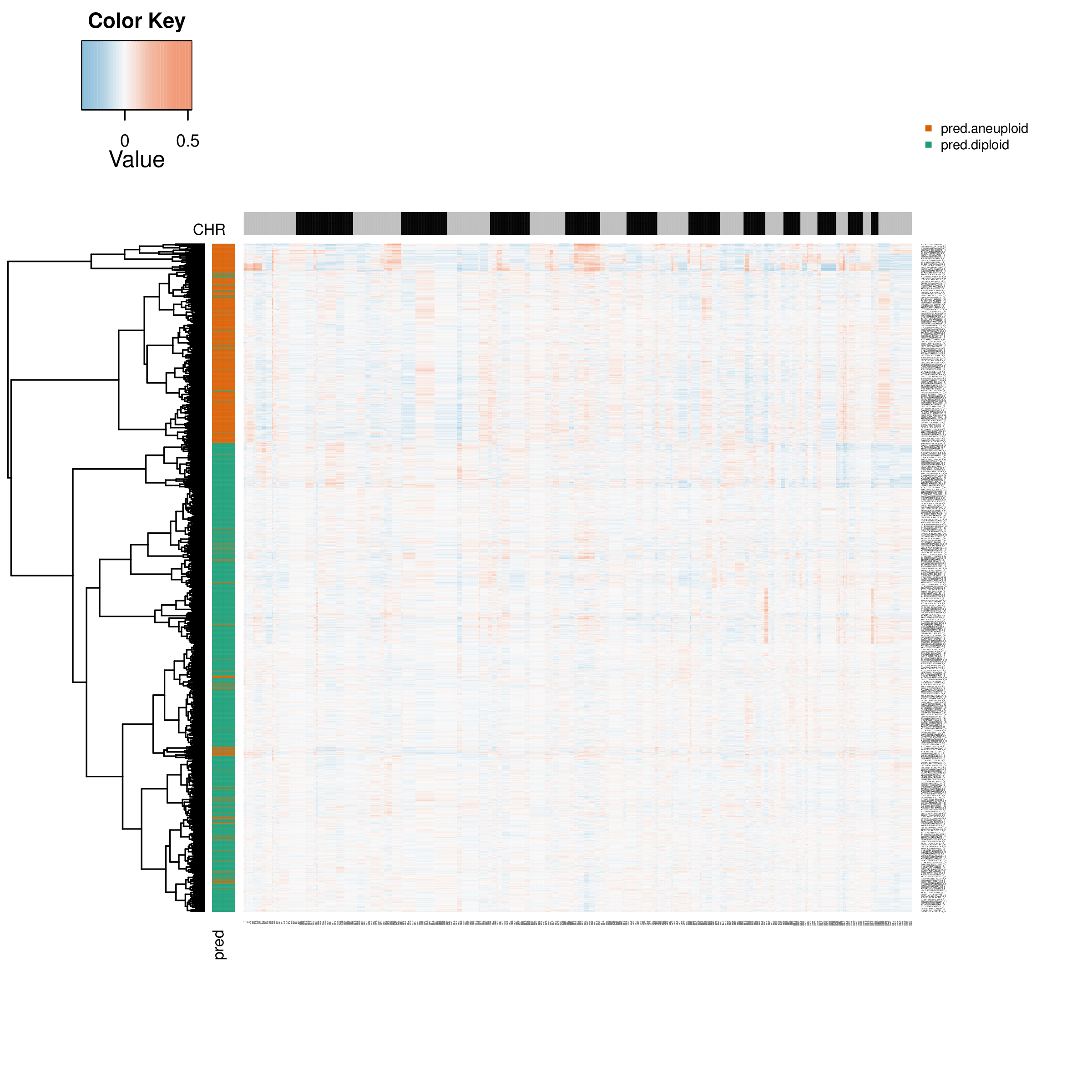
- Graph Interpretation:
- Rows: Represent individual cells.
- Columns: Represent genes sorted by chromosomal position.
- Colors (heatmap body): Red indicates increased copy number, blue indicates decreased copy number.
- Colors (row annotations): The color band on the left is CopyKAT's classification result for cells. Typically, green represents predicted "diploid" (normal) cells, and orange or other warm colors represent predicted "aneuploid" (tumor) cells.
- Analysis Points:
- Observe whether there is an obvious cell population predicted as "aneuploid" that shows clear CNV patterns (large red/blue areas).
- Check whether the cell population predicted as "diploid" has weak CNV signals (colors close to neutral).
Cell Classification Result Dimensionality Reduction Map
To intuitively view the distribution of prediction results in cell space, the report projects the classification results onto a UMAP plot.
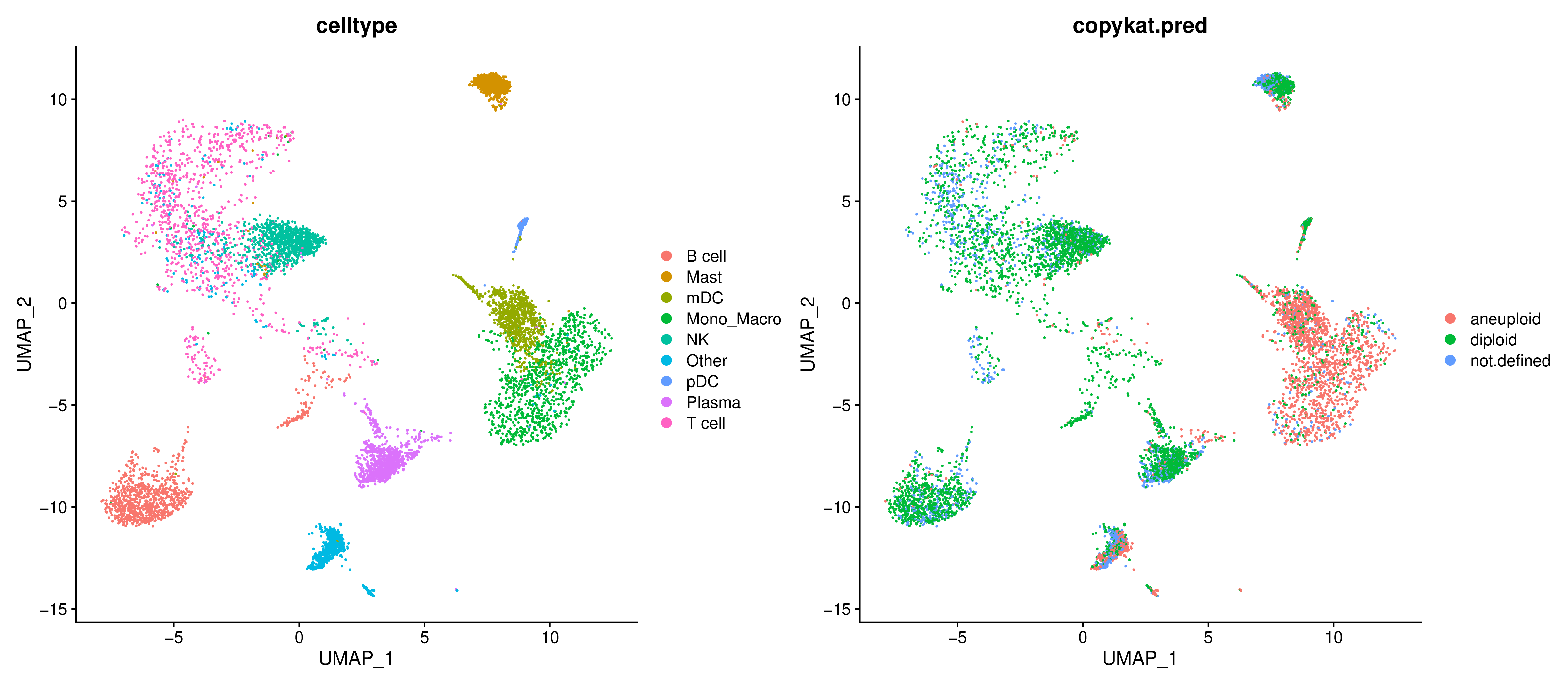
- Graph Interpretation: Each point in the graph is a cell, and the color represents CopyKAT's prediction result (e.g., normal vs. tumor).
- Analysis Points: Check whether the predicted tumor cells highly coincide with your pre-annotated tumor cell clusters.
Tumor Subclone Analysis
After identifying tumor cells, CopyKAT further performs subclone analysis on them.
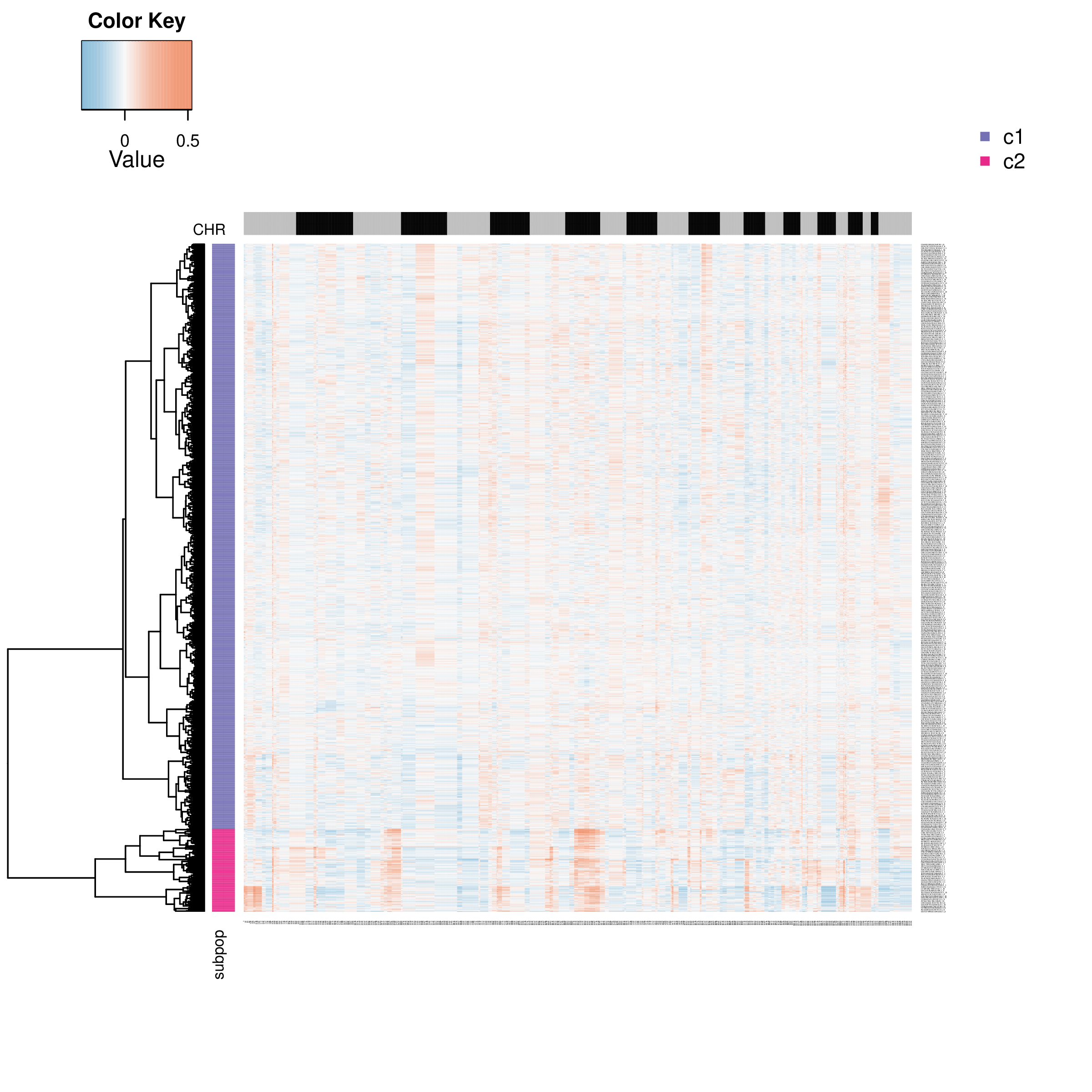
- Graph Interpretation: This heatmap only shows tumor cells predicted as "aneuploid". By clustering their CNV patterns, tumor cells can be divided into different subclones (such as c1, c2 in the row annotations of the heatmap).
- Analysis Points: Compare CNV differences between different subclones. For example, subclone c1 may carry specific chromosomal amplifications that subclone c2 does not, which may represent the evolutionary direction of the tumor.
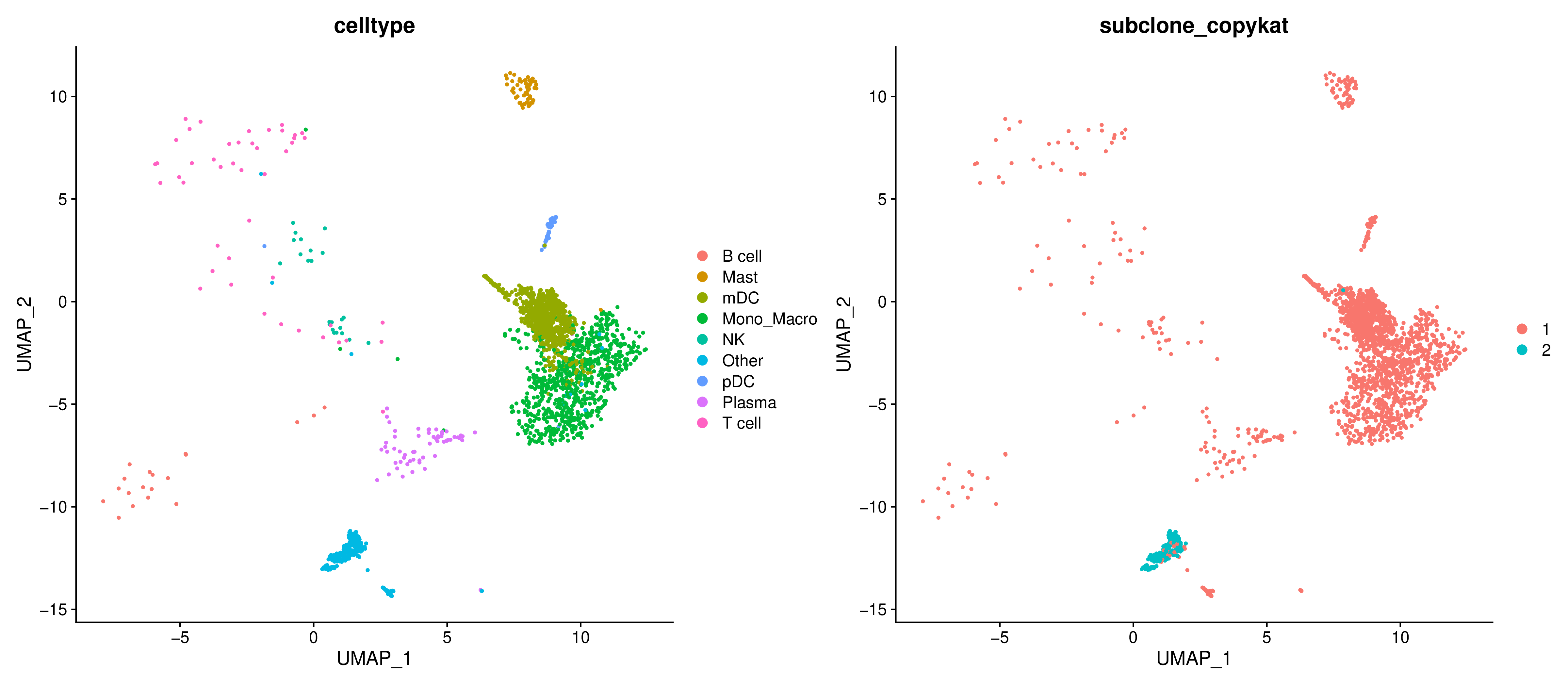
- Graph Interpretation: Projecting the identified tumor subclones (c1, c2, etc.) onto a UMAP plot allows observation of their distribution relationships in cell space.
Application Cases
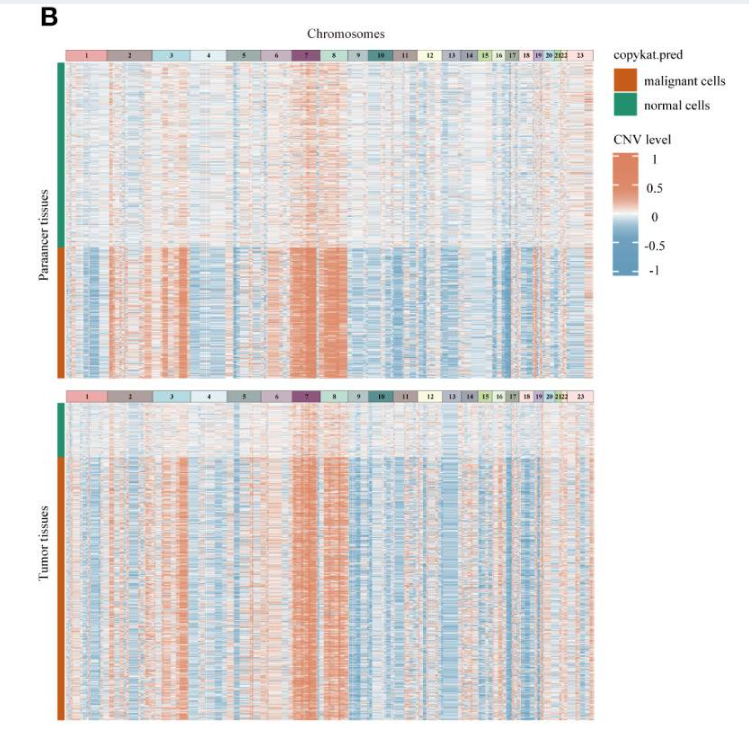
Case 1: Tumor Microenvironment in Osteosarcoma
In a study on undifferentiated pleomorphic sarcoma (UPS), researchers used CopyKAT to distinguish malignant fibroblasts from normal fibroblasts. The results showed that CopyKAT successfully divided fibroblasts into two groups: one with obvious CNV events (malignant) and the other with stable genomes (normal).
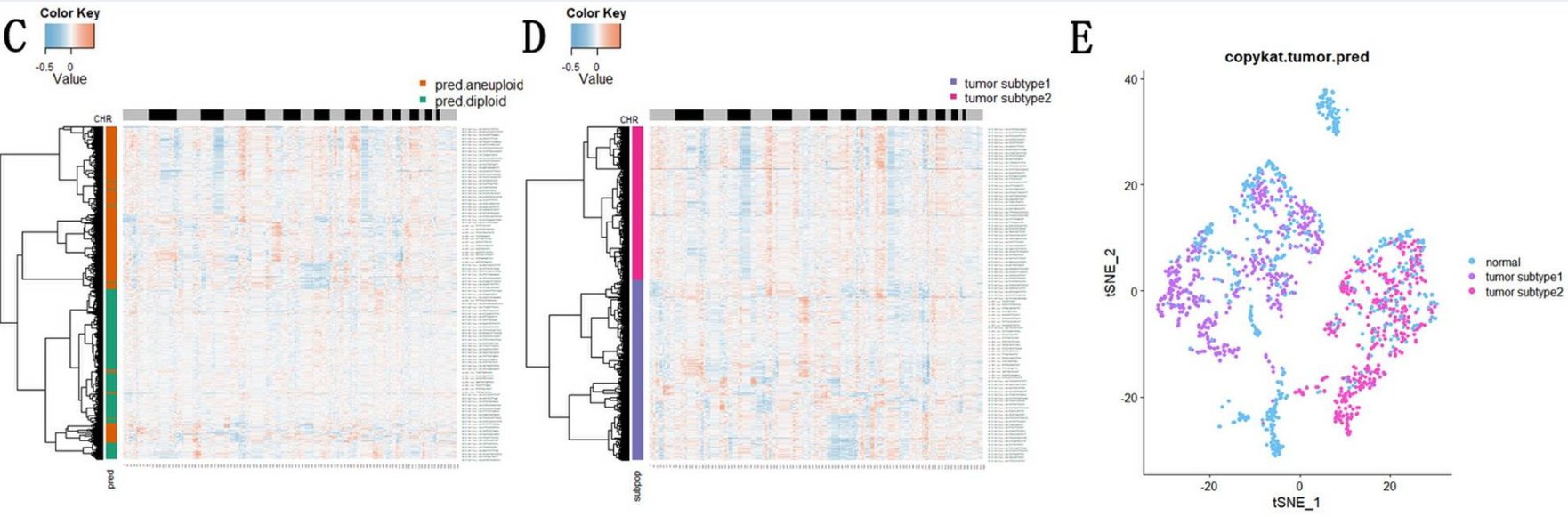
Case 2: Tumor Evolution in Nasopharyngeal Carcinoma
In a study on non-keratinizing nasopharyngeal carcinoma (NPC), CopyKAT was used to identify tumor cells and divide them into two main subclones. Subsequent analysis found that these two subclones had significant differences in metabolism and immune escape-related pathways, revealing functional heterogeneity within the tumor.
Precautions
1. Results Are Not Gold Standard: CopyKAT is a bioinformatics inference based on transcriptomic data, and its classification results of "tumor" or "normal" cells need to be comprehensively judged in combination with other biological evidence (such as key gene markers, other omics data), especially in samples with low tumor purity or weak CNV signals.
2. Quality of Internal Reference Cells: CopyKAT's automatic search for normal diploid cells as internal references is its core advantage, but it may also become a potential risk point. If the cell population to be analyzed does not contain truly normal cells, or the proportion of normal cells is extremely low, the algorithm may incorrectly treat a batch of tumor cells with the least degree of CNV as "normal" references, leading to deviations in the CNV assessment of all subsequent cells.
3. Validation with inferCNV: CopyKAT and inferCNV have different principles and can complement each other. When you are unsure about the results, you can try to use inferCNV (which requires providing clear normal cell references) for cross-validation and observe the consistency of the two results.
Frequently Asked Questions (FAQ)
Q1: What is the difference between copyKAT and inferCNV? Which one should I choose?
A1: The main difference lies in whether manual specification of normal reference cells is required.
- inferCNV: Requires users to explicitly provide a group of "normal" cells as references. Its advantage is that if the reference cells are very reliable, the results will be very accurate. Suitable for cases with clear normal control groups (such as cells from healthy samples, or non-malignant cell types determined to be normal in samples).
- copyKAT: Automatically identifies the most likely normal cells from all cells to be analyzed as internal references. Suitable for scenarios where it is impossible to determine which cells are normal, or when you want to conduct an unbiased malignant/non-malignant exploratory analysis of the entire sample.
- Selection Suggestion: If you have reliable normal references, both can be used, and inferCNV may be more classic; if you do not have reliable normal references, copyKAT is strongly recommended.
Q2: Why are many cells in my results marked as "not defined"?
A2: This usually means that these cells' CNV signals are not clear enough, and the algorithm cannot classify them as "aneuploid" or "diploid" with high confidence. Possible reasons include: low CNV levels in the cells themselves, weak signals due to insufficient sequencing depth, or cells in some intermediate state.
Q3: Can copyKAT be used for non-tumor research?
A3: Theoretically, yes. Any biological scenario that leads to large-scale chromosomal copy number changes, such as certain genetic diseases or genomic instability during cell culture, can be analyzed using copyKAT. However, its algorithm is optimized for tumor scenarios, and interpretation in other scenarios needs to be more cautious.
References
- Gao, R., et al. (2021). Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nature Biotechnology, 39(5), 599-608.
- Yuan, L. L., et al. (2022). Single-cell sequencing reveals the landscape of the tumor microenvironment in a skeletal undifferentiated pleomorphic sarcoma patient. Frontiers in Immunology, 13, 1019870.
- Chen, H., et al. (2023). Comprehensive single-cell transcriptomic and proteomic analysis reveals NK cell exhaustion and unique tumor cell evolutionary trajectory in non-keratinizing nasopharyngeal carcinoma. Journal of Translational Medicine, 21(1), 1-18.